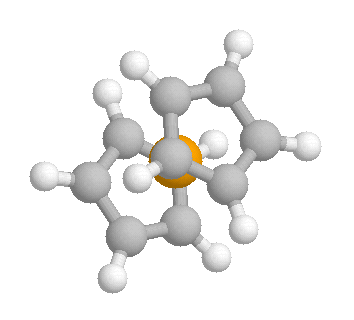
 Why is it that electron diffraction in the gas phase suggests the more
stable
conformation is eclipsed when in the solid state there is seen to be
differently orientated molecules of ferrocene randomly distributed
throughout
the sample.
Why is it that electron diffraction in the gas phase suggests the more
stable
conformation is eclipsed when in the solid state there is seen to be
differently orientated molecules of ferrocene randomly distributed
throughout
the sample.
As I previously posted to the Mad Scientist Network, the rotational barrier in ferrocene is very low but the staggered conformation is the only energy minimum. I was under the apparently mistaken impression that the staggered form was the crystal structure; but you say it's the gas- phase structure.
The hand-waving argument is that imperfections in the molecule-molecule interactions in the crystal give the rings random orientations--or is it (which seems more likely) that even in the crystal, at room temperature, the rings are spinning so that the carbon atoms appear to have a random orientation?
Also, in a model I made using CAChe (Oxford Molecular) I have found the Fe-C distance of an MM-optimised staggered model of ferrocene to be 1.981 Angstrom, but values obtained by electron diffraction (found in texts) of gaseous ferrocene suggest a 2.07 Angstrom distance.
This is not particularly surprising. Molecular mechanics methods are good for approximate geometries, and a difference of 9 picometers between an MM geometry and an experimental geometry is not out of line. I get 2.205 Å for the Fe-C distance from an HF/3-21G* "ab initio" calculation, so your MM method isn't so bad after all.
| Dan Berger |
| Bluffton College |
| http://www.bluffton.edu/~bergerd |